
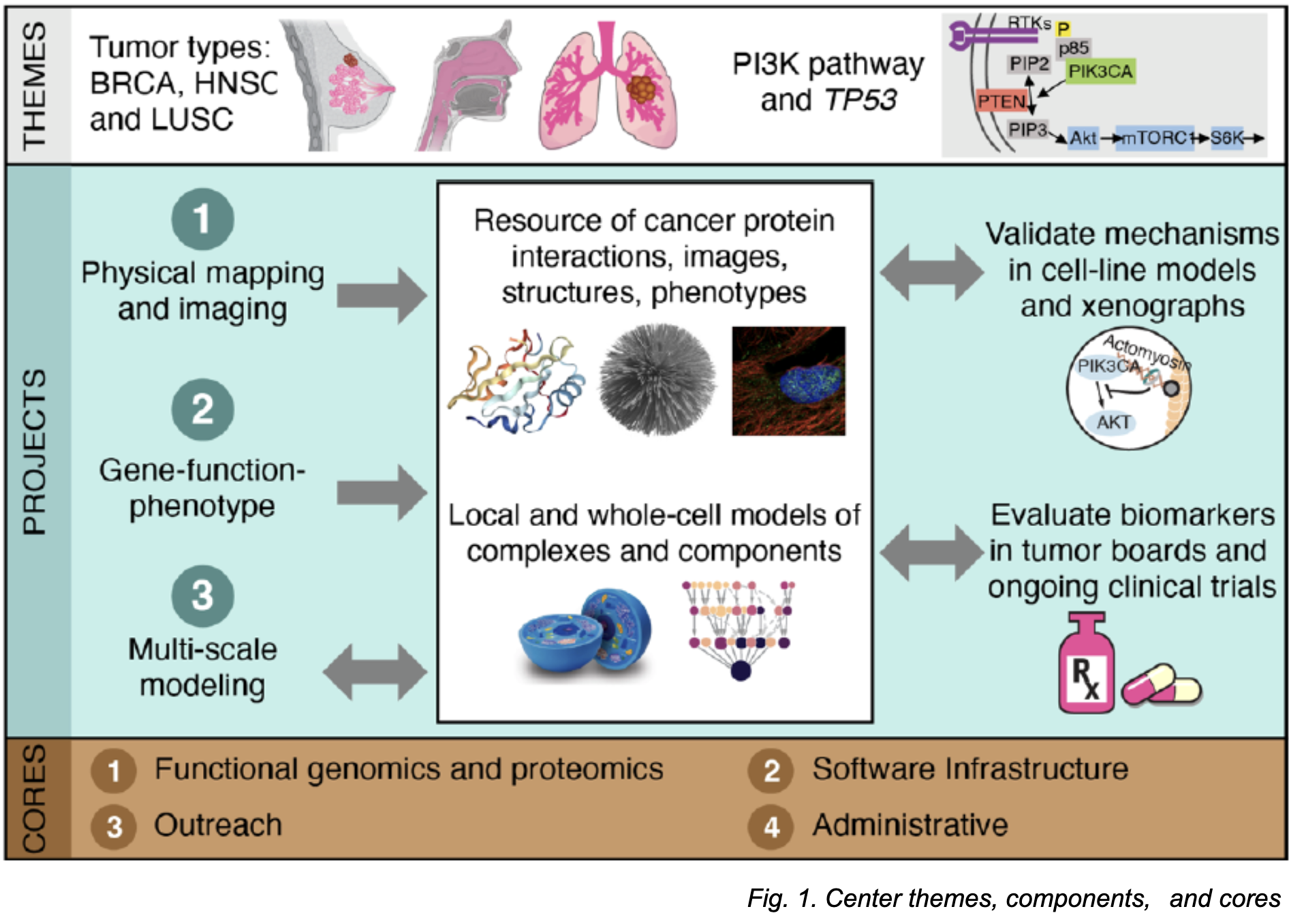
Project 1 performs physical protein mapping and imaging in cancer cell lines [breast (BRCA), head and neck squamous carcinoma (HNSC), and lung squamous cell carcinoma (LUSC). Project 2 studies how gene mutations impact cancer cell function using cell line models and xenografts. Project 3 uses visible neural networks and an Integrative Modeling Platform which determines structures of macromolecular complexes to develop multi-scale maps of protein assemblies in cancer cells.
